■ Installation for Mac OSX
See trouble shooting after the installation.
Download the installer package and install it on your Mac.
If your Mac didn't install R and XQuartz, this installer will automatically download these packages and install them.
■ Basic Usage
Here is sample data
Preparing input file with samtools
・Removing unmapped reads may make the total runtime faster.
$samtools view -b -F 4 inputfile.bam > inputfile.mapped.bam #trimming un-mapped reads
・Type a following command to sort your bam file. (essential)
$samtools sort inputfile.bam inputfile
・Making index file (essential)
$samtools index inputfile.bam
MitoSuite on Mac
(1) Launch MitoSuite.app
(2) Load a bam file from Menu bar | Ctrl+O
If menu bar can not be displayed on your Mac, please use “Ctrl+O” command to load a bam file.
The directory and filename must use only alphanumeric characters.
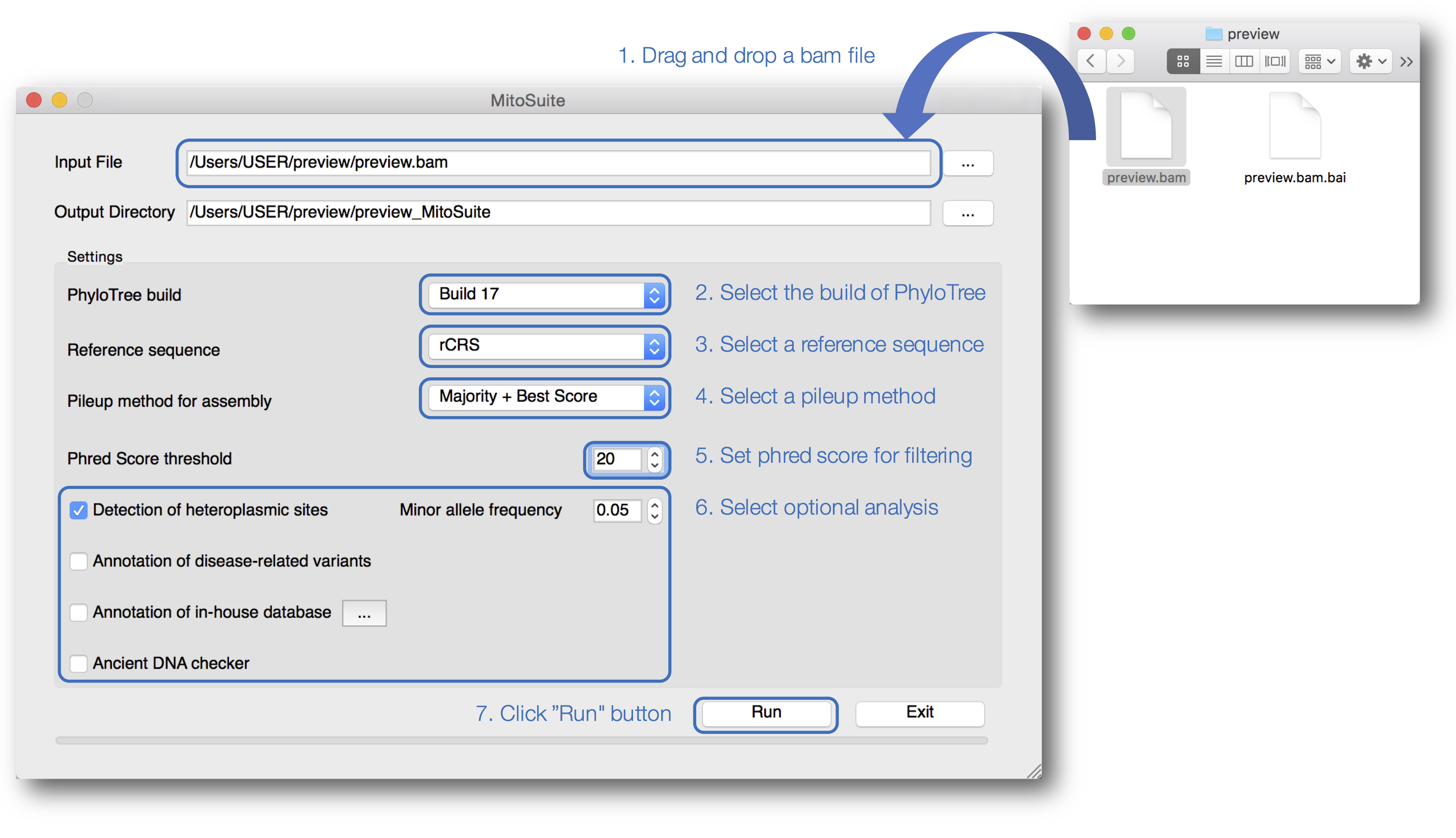
(3) Click “Run” button
■ Automatic installation
See trouble shooting after the installation.
If you use the automated installer, the installation of MitoSuite is very simple.
Build version | Run the automated-installer script (mitosuite_installer.py) in the distribution package.
$python mitosuite_installer.py
Script version | Run the script (MitoSuite_1.0.9b.py) in the distribution package.
$python MitoSuite_1.0.9b.py
If you can not install MitoSuite by this script, please manually install following requirement packages.
After the installation of requirement packages, please run this script again.
■ Manual installation
Requirement packages for MitoSuite can also be installed manually.
[1] Install R (with "ggplot2", "reshape2" packages)
Please install R on your Linux according to r-project.org.
Open your terminal.app and type a following command to start R console.
$R
Then, install "lazyeval","ggplot2" and "reshape2" packages in R console.
Depending on the version of R, ggplot2's bugs caused by the namespace of lazyeval are reported (https://blog.rstudio.org/2016/11/14/ggplot2-2-2-0/).
This bug is resolved by installing the latest version of the lazyeval package.
>install.packages(c("lazyeval",“ggplot2”,"reshape2"),dep=T)
>q()
[2] Install Python modules
You can easily install requirement modules using the following one-line command.
$sudo pip install numpy scipy pandas 'pysam >=0.8.4,<0.11.0' pyinstaller -U
Please install PySide based on your Linux system.
Ubuntu, Debian:
$sudo apt-get install python-pyside
CentOS, Fedora, RedHat and RPM-based (as root):
$sudo yum install python-pyside pyside-tools
OpenSUSE:
$sudo zypper ar http://download.opensuse.org/repositories/home:/cgoncalves:/pyside:/ shiboken/openSUSE_11.2/ pyside
$sudo zypper refresh pyside
$sudo zypper install python-pyside
If you can not install PySide by these procedures, please check these pages (https://wiki.qt.io/PySide_Binaries_Linux
or http://pythoncentral.io/install-pyside-pyqt-on-windows-mac-linux/or http://pyside.readthedocs.io/en/latest/building/linux.html).
■ Basic Usage
Here is sample data
Preparing input file with samtools
・Removing unmapped reads may make the total runtime faster.
$samtools view -b -F 4 inputfile.bam > inputfile.mapped.bam #trimming un-mapped reads
・Type a following command to sort your bam file. (essential)
$samtools sort inputfile.bam inputfile
・Making index file (essential)
$samtools index inputfile.bam
MitoSuite on Linux
(1) Launch MitoSuite via terminal
Build version |
$ mitosuite
Script version |
$ python MitoSuite_1.0.9b.py
(2) Load a bam file from Menu bar | Ctrl+O
The directory and filename must use only alphanumeric characters.
(3) Click “Run” button
- Phred Score threshold
Threshold value shows the lower of the base-call quality (phred score) to call consensus bases.
- Pileup method for calling consensus bases
Majority : Deciding the base by the majority of the number of basecall
Example 1 | Total depth at a site = 30 ("A":28, "G":2) → Consensus base : "A"
Example 2 | Total depth at a site = 30 ("A":15, "G":15) → Consensus base : "N"
BestScore : Deciding the base with the highest basecall quality at each site
Example 1 | Total depth at a site = 10 ("A":8, "G":2), Best phred score at each bases : "A"(Q=35), "G"(Q=40) → Consensus base : "G"
Example 2 | Total depth at a site = 10 ("A":8, "G":2), Best phred score at each bases : "A"(Q=40), "G"(Q=40) → Consensus base : "N"
Majority + BestScore : Incorporating the best quality method with the majority decision method at each site
Example 1 | Total depth at a site = 10 ("A":5, "G":5), Best phred score at each bases : "A"(Q=35), "G"(Q=40) → Consensus base : "G"
Example 2 | Total depth at a site = 10 ("A":5, "G":4), Best phred score at each bases : "A"(Q=40), "G"(Q=40) → Consensus base : "A"
[1] Interactive graphs are not drawn on Firefox (ver.67 or later).
1. Enter "about:config" in the Firefox address bar.
2. Change the value of “security.fileuri.strict_origin_policy” from true to false.
Note: This procedure can change the security level of your web browser. After using MitoSuite, please return to the default setting (false to true).
[2] SVG files are not generated.
1. Download ggplot2 ver.2.2.1 (less than 3) and install from R.
$curl -O https://cran.r-project.org/src/contrib/Archive/ggplot2/ggplot2_2.2.1.tar.gz
or
$wget https://cran.r-project.org/src/contrib/Archive/ggplot2/ggplot2_2.2.1.tar.gz
$R CMD INSTALL ggplot2_2.2.1.tar.gz
Mac OSX | Download the installer package and install it on your Mac.
Linux (Build version) | Download the package & run the installer script on your terminal.
Linux (Script version) | Download the package & run the script on your terminal.
Load a bam file and click a "Run" button
Here is sample data
Go to an output folder (/inputfile_MitoSuite)
View analysis results (results.html) through a web browser
※Recommended web browsers | Firefox or Safari
Here is an example output
Comprehensive mitogenome analysis for high-throughput sequencing
Here is an example output
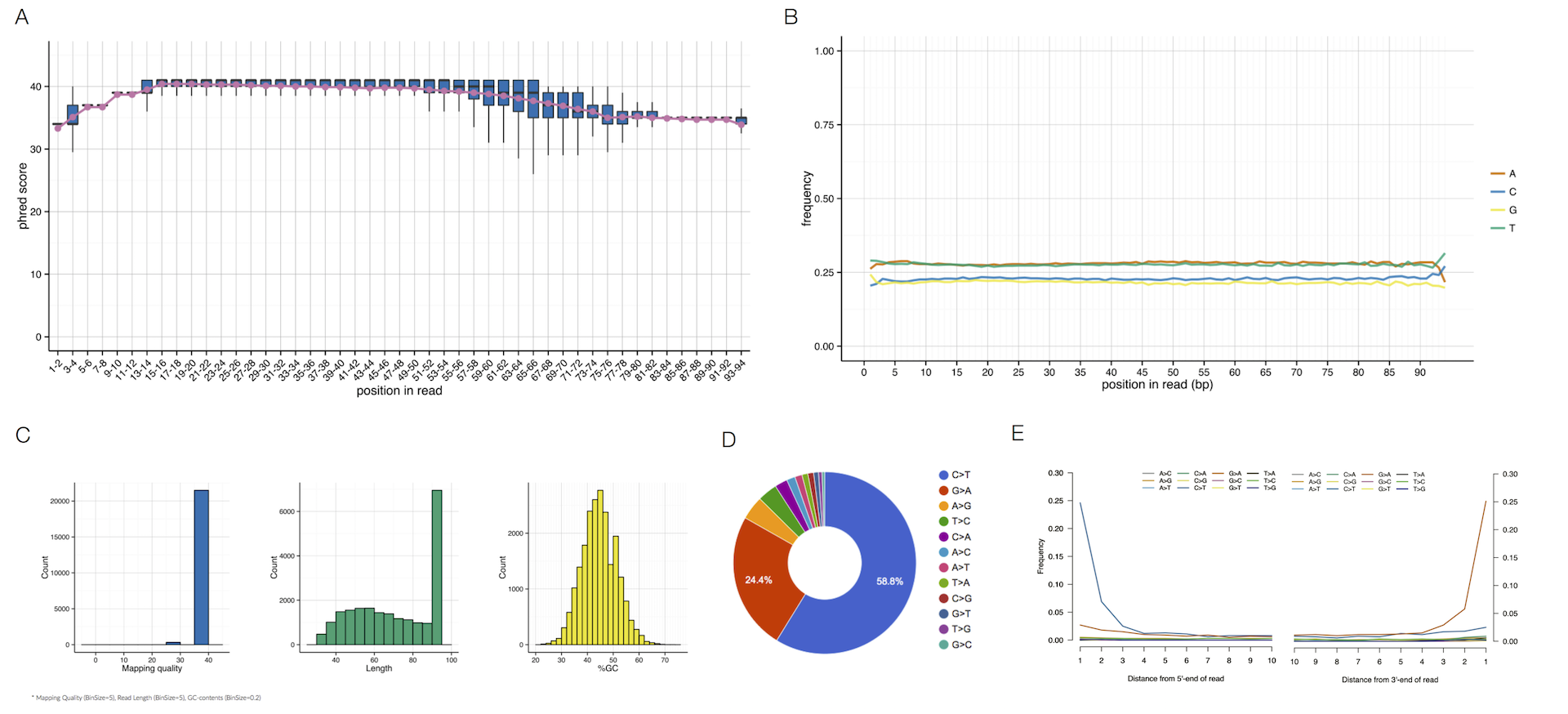
MitoSuite can fully support Bam files without any file conversions. This tool can easily check the quality of NGS data by directly parsing a Bam file.
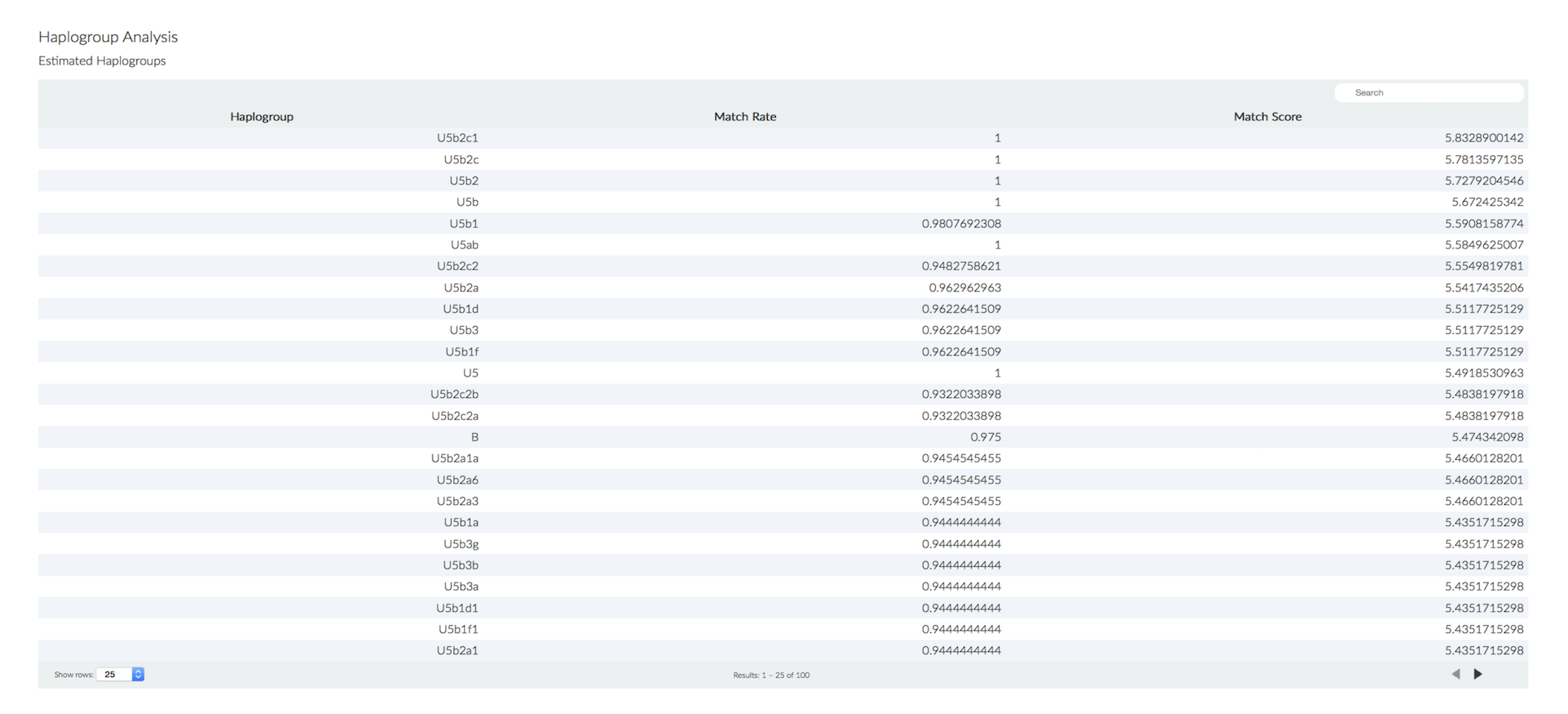
MitoSuite can easily get genetic profiles about mitochondrial genome. These profiles are visualized and users can see them through a web browser.
Test environment | Mac OSX 10.7 - 10.11
Latest version | MitoSuite_1.0.9.dmg, CUI (beta)
Test environment | Ubuntu 12.04 (32 bit)
Latest version | Build version, Script version
Bam files + Output files + Template file | sample_data.tar.gz
Template file | template.csv
This template file is supported by MitoSuite's version >= 1.0.5. The annotation file is a common comma-delimited CSV format (.csv) containing two items: a mutation allele with a genomic position corresponding to that of rCRS (e.g., C150T, A4282G), and related information (e.g., related-disease name) in each designated column. MitoSuite can parse the in-house annotation data (.csv), and then annotate variants according to the file.
Please use the latest bug-fixed version.
Version 1.0.9 | Bug fixes (6 June, 2017)
Version 1.0.8 | Minor fixes of coverage calculation, Performance improvements, MitoSuite for Linux supports pysam > v.0.8.4 except for the the latest version (3 May, 2017)
Version 1.0.7 | Performance improvements and bug fixes related to user experience (1 Apr., 2017)
Version 1.0.6 | Performance improvements and bug fixes related to user experience (29 Mar., 2017)
Version 1.0.5 | Support for customizable (in-house) annotation data, Removed version limits in all dependent python packages (Linux),
Release of the build and script version (beta) for Linux, Improvements in graphical interface and installation, Bug fixes (20 Mar., 2017)
Version 1.0.4 | Bug fixes (31 Dec., 2016)
Version 1.0.3 | Bug fixes (14 Oct., 2016)
Version 1.0.2 | Improvements in graphical interface and installation, Support for Phylotree Build 17, Bug fixes (25 Aug., 2016)
Version 1.0.1 | Bug fixes (20 Apr., 2016)